
Abstract
A phase 2, open-label study in 21 patients with non-Val30Met and non-Val122Ile hereditary transthyretin amyloidosis showed that tafamidis (20 mg daily for 12 months) stabilized these transthyretin variants. We assessed cardiac amyloid infiltration and cardiac abnormalities in this same study population. At baseline, median age was 64.3 years, 11 patients were in NYHA class II, 13 had conduction abnormalities, 14 N-terminal pro-hormone brain natriuretic peptide concentrations >300 pg/ml, and 17 interventricular septal thickness >12 mm. Mean (SD) left ventricular ejection fraction was 60.3 % (9.96). Patients with normal heart rate variability increased from 4/19 at baseline to 8/19 at month 12 (p < 0.05). Cardiac biomarkers remained stable. Although four patients had increases in interventricular septal thickness ≥2 mm, the remainder had stable septal wall thickness. There were no clinically relevant changes in mean echocardiographic/electrocardiographic variables and no safety concerns.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hereditary transthyretin (TTR) amyloidosis is a rare progressive disease dominated by cardiomyopathy and/or polyneuropathy [1, 2]. The pathophysiology of this disorder involves a mutation in the TTR gene that destabilizes the tetrameric TTR protein, resulting in accelerated tetramer dissociation and misfolded monomers that aggregate into insoluble amyloid fibrils. These fibrils infiltrate the myocardium and cardiac conduction system or peripheral and autonomic nerves and can also build up in other organs (e.g., gastrointestinal tract, kidneys, and eyes) [1, 2]. They disrupt normal tissue structure and lead to progressive organ failure [1, 2]. TTR cardiomyopathy can also occur in the absence of genetic mutations, often in elderly males (wild-type TTR amyloidosis or senile systemic amyloidosis) [1, 3].
Hereditary TTR amyloidosis is a highly heterogeneous disease associated with over 100 mutations of the TTR gene [4]. Penetrance and severity of disease manifestations are highly variable, and the typical clinical picture and course of disease of different TTR mutations are only beginning to be defined. Certain mutations produce a primarily neurological phenotype (e.g., Val30Met; [5, 2]), others are associated almost exclusively with cardiac problems (e.g., Val122Ile; [6]), whereas numerous other mutations induce mixed phenotypes.
Polyneuropathy associated with TTR amyloidosis is typically characterized by small fiber, sensory-motor neuropathy involving symptoms such as pain, sensory loss, numbness, and motor deficits, including walking difficulties [2]. Once such neuropathy symptoms appear, progressive autonomic or cardiac dysfunction leads to death within an average of 10 years [2].
Cardiac manifestations in TTR amyloidosis can range from asymptomatic arrhythmias to severe restrictive cardiomyopathy with heart failure [7, 8, 1]. Cardiomyopathy is the dominant clinical manifestation in patients harboring the aforementioned Val122Ile mutation or Thr60Ala, Ile68Leu, Ser77Tyr, and Leu111Met mutations [9, 1, 3]. Patients with considerable cardiac abnormalities tend to be older at the onset of disease and have reduced survival rates thereafter [3].
Because TTR is produced primarily in the liver, transplantation of a liver with wild-type TTR genes can be an effective therapy, particularly in patients with Val30Met mutations, and when performed early in the disease before extensive amyloid deposits have accumulated [10, 11, 2]. However, liver transplantation is associated with inherent risks, particularly those accompanying chronic immunosuppression [10, 12]. Furthermore, it frequently fails to prevent the progression of cardiomyopathy and conduction disturbances [13, 14, 12], probably due to deposition of wild-type TTR onto pre-existing amyloid deposits [15, 2]. This may partially explain the reduced 5-year survival rates after liver transplantation reported for non-Val30Met patients [16] and has led surgeons to consider combined liver and heart transplantation in certain cases [1].
Pharmacologic therapy of hereditary TTR amyloidosis has become a possibility with the development of TTR stabilizers, which target the rate-limiting step in the formation of amyloid fibrils [17, 18]. By specifically binding to the tetramers, TTR stabilizers increase tetramer stability and inhibit their dissociation into amyloidogenic monomers [19, 18]. One example is diflunisal, a generic nonsteroidal anti-inflammatory drug that was found to reduce the rate of progression of polyneuropathy and to preserve quality of life in patients with TTR familial amyloid polyneuropathy [20]. However, nonsteroidal anti-inflammatory drugs can cause sodium retention and renal dysfunction, limiting or preventing the use of diflunisal in subjects with cardiomyopathy or heart failure. Another TTR stabilizer is tafamidis meglumine [21] that was shown in an 18-month, randomized, controlled, clinical trial to slow the progression of peripheral neurologic impairment in patients with TTR amyloid polyneuropathy carrying the Val30Met mutation [22]. We recently reported on the results of a subsequent open-label study examining the effects of tafamidis in patients with non-Val30Met TTR mutations [23]. This study demonstrated that tafamidis was well tolerated and effectively stabilized tetrameric TTR in this TTR amyloid polyneuropathy patient population [23]. Furthermore, exploratory efficacy results indicate that the utility of tafamidis observed in Val30Met patients may extend to patients with TTR amyloidosis, regardless of the underlying TTR mutation [23, 24].
Considering that cardiac amyloidosis and electrocardiographic and echocardiographic changes tend to progress despite liver transplantation [13, 15, 25] and, thereby, severely limit the benefits of this procedure in patients with cardiac manifestations, it is of particular importance to determine whether the observed efficacy of tafamidis on neurological symptoms also extends to cardiac outcomes. Thus, the current post hoc analysis examined safety and efficacy of tafamidis with respect to cardiac structure and function in the previously described [23] study population of non-Val30Met and non-Val122Ile patients with TTR amyloidosis.
Methods
Patients, Study Design, and Outcomes
As described previously [23], this 12-month, open-label study was conducted at four research centers in France, Germany, Italy, and the USA from June 2008 to January 2010. Eligible patients were men and non-pregnant women 18 to 75 years of age who had symptomatic, biopsy-confirmed TTR amyloid polyneuropathy (i.e., peripheral neuropathy or autonomic neuropathy) due to a known pathogenic TTR mutation other than the previously studied Val30Met mutation [22] or the Val122Ile mutation assessed in a separate study (clinical trial identifier: NCT00694161). All patients received tafamidis 20 mg (in a soft gelatin capsule) once daily.
Besides evaluating TTR stabilization, safety and tolerability of tafamidis meglumine, and changes in neurological function, this study also examined exploratory efficacy outcomes to assess pre-treatment cardiac involvement at baseline and the effects of tafamidis treatment on cardiac function at month 6 and month 12. These exploratory outcomes included 12-lead electrocardiography, 24-h Holter monitoring, heart rate response to deep breathing (HRDB), cardiac biomarkers (N-terminal pro-hormone brain natriuretic peptide (NT-proBNP) and troponin I), and echocardiography. In addition to the standard evaluations at baseline, month 6, and month 12, electrocardiography and biomarker evaluations were also performed at weeks 2, week 6, and month 3. For consistency of interpretation, electrocardiograms, echocardiograms, and 24-h Holter monitoring results were read at a central laboratory. To assess HRDB, the average heart rate difference was measured while patients took a series of eight deep breaths at the optimal rate for maximum heart rate variability (i.e., six breaths per minute) and then compared with normative values.
Statistical Analyses
The safety and intent-to-treat populations consisted of all enrolled patients who received ≥1 dose of tafamidis. Analyses were performed based on observed data from the intent-to-treat population. For the exploratory efficacy end points, the observed values and change from baseline over time for continuous variables as well as treatment-emergent categorical abnormalities were summarized at each follow-up time point by descriptive statistics.
Categorical echocardiographic abnormalities included: left ventricular (LV) posterior or septal wall thickness >12 mm (primary echocardiographic evidence for myocardial amyloidosis [26]), LV ejection fraction (LVEF) <50 %, ratio of early (E) over late (atrial; A) peak diastolic mitral inflow velocity (E/A ratio) ≥2, ratio of E over the early diastolic mitral annular velocity (e′; E/e′ ratio) >15 (evidence of elevated LV filling pressure), E deceleration time ≤150 ms, isovolumic relaxation time ≤70 ms, abnormal regional wall motion valve thickening, valvular regurgitation, right ventricular thickness ≥7 mm, abnormal respiratory variation of inferior vena cava (evidence of elevated right ventricular filling pressure), and pericardial effusion.
Treatment-emergent Holter monitoring abnormalities included atrial fibrillation/flutter, atrial tachycardia, non-sustained ventricular tachycardia (<30 beats), sustained ventricular tachycardia (≥30 beats), sinus pause, and heart rate variability (HRV). HRV indices included the percentage of successive RR intervals with >50 ms difference between beats (pNN50), the root mean square of successive differences of the RR intervals (ms) between normal beats (RMS-SD), the Magid standard deviation of all RR intervals (ms) for all 5-min segments, and the Kleiger standard deviation of all RR intervals.
We undertook post hoc analyses to evaluate changes from baseline to month 12 in echocardiographic measures of myocardial amyloid infiltration, LV systolic dysfunction, and LV diastolic dysfunction. To assess cardiac amyloid infiltration during tafamidis treatment, we used the International Society of Amyloidosis’ evaluation criterion for immunoglobulin light-chain amyloidosis that defines disease progression as increases in IVS ≥2 mm compared with baseline [26]. Furthermore, in the absence of a direct comparator group, we decided to use a 10 % change approach for assessing changes in LV mass during the treatment period, with a ≥10 % increase classified as progressive amyloid infiltration, any change <10 % as stabilized disease, and a ≥10 % decrease as regression. Similarly, using LVEF as an estimate of systolic function, we classified a ≥10 % increase in LVEF as an improvement, a ≥10 % decrease as a deterioration, and any change <10 % as preserved functional capacity. Finally, to gain some insight into changes in diastolic function and filling pressure during tafamidis treatment, all patients with evaluable assessments were categorized into one of three groups based on their lateral E/e′ ratio (a parameter known to be correlated to left ventricle filling pressure): group A if E/e′ <8 (low filling pressure), group B if E/e′ is between 8 and 15, and group C if E/e′ >15 (high filling pressure). This standard categorization [27] was performed at baseline, month 6, and month 12.
Results
Patient Disposition and Baseline Characteristics
A total of 21 patients were enrolled and received ≥1 dose of tafamidis. Two patients who were over 75 years of age but met all other eligibility criteria were granted waivers and enrolled in the study.
Baseline demographic and clinical characteristics of the study population have been reported previously [23] and included significant neurologic deficits and impaired disease-specific quality of life (Table 1). Looking at TTR amyloidosis-related medical history events, all patients reported symptoms of peripheral or autonomic neuropathy at study entry. In addition, 13/21 patients (61.9 %) also reported symptoms attributable to cardiomyopathy prior to their enrollment. At baseline, 10/21 patients (47.6 %) were in New York Heart Association class I and 11 (52.4 %) in class II, and four patients (19.0 %) had cardiac pacemakers prior to enrollment.
Safety Findings
Tafamidis was well tolerated. Eighteen (85.7 %) patients completed the study and three (14.3 %) discontinued early, one because of a transient ischemic attack and two to undergo liver transplant. No life-threatening adverse events or patient deaths were reported. Of the eight (38.1 %) patients reporting serious treatment-emergent adverse events, five experienced falls and three experienced one serious cardiovascular adverse event each (one coronary artery stenosis, one transient ischemic attack, and one atrioventricular block). The patient with the transient ischemic attack withdrew from the study (Thr60Ala, history of hypertension); the other two were treated by implantation of a new stent at a site of in-stent restenosis or of a pacemaker/cardioverter-defibrillator respectively and recovered without any change in study drug.
Electrographic analysis of heart rate and HRV parameters demonstrated no adverse effects of tafamidis treatment on cardiac conduction or repolarization; only one patient had a change to >500 ms in QTcB (corrected QT interval calculated using Bazett’s heart rate correction formula), and no patient developed an increase ≥60 ms in QTcF (corrected QT interval calculated using Fridericia’s correction method) while receiving tafamidis.
Electrocardiographic Findings
Mean heart rate and mean electrocardiography intervals were within normal ranges at baseline. The mean (SD) QRS was slightly prolonged at 101.8 (21.4) ms but below the abnormal value of 120 ms. No relevant changes were observed over the 12-month study period in mean electrocardiographic measurements (Table 2).
At baseline, 16/21 patients (76.2 %) had electrocardiographic abnormalities (Table 3) [23]. Most frequent were conduction abnormalities (13/21, 61.9 % of patients) such as left anterior hemiblock and first-degree atrioventricular block [23]. During the 12-month study period, 9/21 patients (42.9 %) developed new-onset, treatment-emergent electrocardiographic abnormalities; these were predominantly arrhythmias (8/18, 44.4 %). However, the proportion of patients with electrocardiographic abnormalities at month 12 (13/18, 72.2 %) did not exceed that at baseline.
Holter Monitoring Findings
At baseline, heart rate assessments were normal (mean [SD] maximum heart rate, 110.9 [15.2] bpm; mean 24-h heart rate, 74.7 [12.1] bpm; minimum heart rate, 55.1 [10.7] bpm), and there were no clinically relevant changes in these parameters during the study period.
Holter monitoring abnormalities were seen in 14/21 patients (66.7 %) at baseline; most common were atrial tachycardia (11/21, 52.4 %) and non-sustained ventricular tachycardia (8/21, 38.1 %) (Table 2). New, treatment-emergent abnormalities on Holter monitoring were seen in 9/19 patients (47.4 %); most common were atrial tachycardia and non-sustained ventricular tachycardia (4/21, 19 % each).
Regarding HRV assessments at baseline, mean Kleiger and Magid standard deviation values were abnormal (≤100 ms), but RMS-SD and pNN50 were within normal range (≥30 ms and ≥10 %, respectively) (Fig. 1). Mean values for the HRV indices did not deteriorate over the study period, and the number of patients with normal HRV (defined as normal pNN50, RMS-SD, and Kleiger SD) in the safety population increased from 4/19 (21 %) at baseline to 8/19 (42 %) at month 12 (p = 0.0455; Fig. 2).

Holter monitoring HRV values in the safety population showed that mean and median values for the percentage of successive RR intervals with >50 mg difference between normal beats (pNN50) (a), the root mean square of successive differences of the RR intervals between normal beats (RMS-SD) (b), the Magid SD (c), and the Kleiger SD of all RR intervals (d) did not deteriorate over the study period. Bars indicate the range of values observed, boxes span from the 25th to the 75th percentiles, horizontal lines within the boxes indicate median change, and plus sign indicates mean changes. HRV heart rate variability, SD standard deviation

The proportion of patients with normal HRV in the safety population increased from 4/19 at baseline to 8/19 at month 12 (p < 0.05). p values are based on two-sided McNemar’s tests. HRV heart rate variability, ns non-significant
Heart Rate Response to Deep Breathing
HRDB is a non-invasive measure of autonomic function and vagal cardiac response. The mean normal deviate for HRDB in the population of patients with evaluable assessments was –0.7 at baseline and remained stable over time (change from baseline at month 12 = –0.1) (Table 4). However, only 12/21 patients had an evaluable assessment at baseline, and of these, only 7 also had an evaluable assessment at month 12.
Cardiac Biomarkers
The mean NT-proBNP concentration was elevated at baseline, and no clinically relevant changes were noted during the study [23]. There were 14 patients (66.7 %) with an NT-proBNP concentration >300 pg/ml at baseline, 10 of whom had concentrations >1000 pg/ml. However, with the exception of one patient whose NT-proBNP concentration steadily increased from 2603 pg/ml at baseline to 7902 pg/ml at month 12, there were no consistent clinically meaningful increases above each individual’s baseline concentration during the 12-month tafamidis treatment (Fig. 3).

NT-proBNP concentrations in individual patients with baseline concentrations <300 pg/ml (a) or >300 pg/ml (b). No consistent, clinically meaningful increases in NT-proBNP were detected during 12 months of tafamidis treatment, with the exception of a steady rise from 2603 to 7902 pg/ml in patient 10. NT-proBNP N-terminal pro-hormone brain natriuretic peptide. a Patients had missing baseline data, and the last pre-treatment assessment during screening is depicted. b Patient discontinued tafamidis on day 31 following a transient ischemic attack. c Patient discontinued tafamidis prior to month 12 to undergo combined liver/heart transplant. d Patient discontinued tafamidis prior to month 3 to undergo a liver transplant
As previously reported, mean troponin I concentrations were within normal ranges (≤0.1 ng/ml) at baseline and remained stable during tafamidis treatment [23]. Only one patient had abnormal troponin I levels at baseline (0.134 ng/ml at baseline and 0.117 ng/ml at month 12), two patients developed abnormal levels during the study (one had <0.050 ng/ml at baseline and 0.130 ng/ml at months 6 and 12, the other had <0.050 ng/ml from baseline to month 6 and <0.150 ng/ml at month 12), and another study completer had one abnormal reading at week 6 (0.110 ng/ml).
Echocardiographic Findings
Several echocardiographic measures were abnormal at baseline including the mean (SD) values for LV posterior wall thickness (LVPW) of 14.6 (2.6) mm [23], interventricular septal thickness (IVS) of 15.2 (2.8) mm [23], isovolumetric relaxation time of 84.1 (17.5) ms, and E/e′ lateral ratio of 13.1 (7.8). Despite this evidence of cardiac amyloidosis at baseline, average echocardiographic values remained stable during the 12-month study period [23].
As previously described [23], all patients had at least one pre-specified echocardiographic abnormality at baseline. The most frequent echocardiographic aberrations were increased LVPW and increased IVS (17/19, 89.5 % each) closely followed by valve thickening (16/19, 84.2 %). In addition, 8 of 14 patients (57.1 %) had evidence of an elevated LV filling pressure, 5 of 16 patients (31.3 %) reported elevated right ventricular filling pressures, and 3 of 21 patients (14.3 %) had an ejection fraction <50 % (Table 5). During the 12-month study period, new-onset echocardiographic abnormalities were reported in 12 of 19 patients (63.2 %), the most common being LVPW >12 mm (2/3, 66.7 %), valvular thickening (3/5, 60 %), and thickening of the right ventricular wall to ≥7 mm (6/13, 46.2 %).
At baseline, 17 of 19 patients with an evaluable assessment (89.5 %) had left ventricular wall thickening with IVS and LVPW of >12 mm, demonstrating amyloid infiltration of the myocardium. At the end of the study, left ventricular wall thickness measures remained below >12 mm threshold in the two patients with normal IVS at baseline. As previously reported [23], 4 of the 12 patients (33.3 %) with evidence of cardiac amyloid at baseline and a valid assessment at month 12 demonstrated ≥2 mm increases in IVS thickness (range, 2–6 mm), indicative of modest disease progression during the 12-month treatment period. Moreover, of the 14 patients with available measurements, six (42.9 %) had a ≥10 % increase in LV mass at month 12 whereas LV mass was stable in seven (50.0 %) and decreased by >10 % from 228 to 201 g in the 14th patient (Fig. 4).

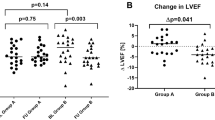
Changes in cardiac amyloid infiltration, systolic function, and LV filling pressure from baseline within individual patients. a LV mass, as a measure of cardiac amyloid infiltration, was maintained or improved (<10 % increase) in 8/14 (57.1 %) from baseline to month 12. b LVEF as a measure of systolic function was maintained or improved (<10 % decrease) in 16/18 (88.9 %) from baseline to month 12. c Lateral E/e′ as an estimate of LV filling pressure was normal (E/e′ <8), undetermined (8 ≤ E/e′ ≤ 15), or elevated (E/e′ > 15) in 6/16 (37.5 %), 5/16 (31.3 %), and 5/16 (31.3 %) at baseline and in 3/11 (27.3 %), 5/11 (45.5 %), and 3/11 (27.3 %) at month 12, respectively. LV left ventricular, LVEF LV ejection fraction, E/e′ ratio of peak mitral inflow velocity of early filling to early diastolic mitral annular velocity
Using LVEF to estimate LV systolic function, apart from temporary decreases in LVEF to 42 and 37 % in two patients at month 6, none of the 17 patients with an LVEF of ≥50 % at baseline developed LV systolic dysfunction (LVEF <50 %) during the 12-month tafamidis treatment. Furthermore, a post hoc analysis showed that LVEF deteriorated by ≥10 % in two (11.1 %) of the 18 study completers, but was maintained (n = 14, 77.8 %) or increased (n = 2, 11.1 %) at the end of the 12-month study in the others.
At baseline, left ventricular filling pressure, determined from the lateral E/e′ ratio, was within the normal range in 6/16 (37.5 %) patients with available data, but was undetermined in 5/16 (31.3 %) and elevated in another 5/16 (31.3 %) patients. This distribution did not change markedly during tafamidis treatment, with a small shift from normal to undetermined.
Discussion
Previously reported findings from this open-label phase 2 study showed that tafamidis 20 mg once daily was well tolerated over 12 months in patients with non-Val30Met and non-Val122Ile TTR amyloidosis with some worsening in neurological manifestations and maintenance of health-related quality of life and nutritional status [23]. This analysis extends these results by showing that, overall, there was no clinically meaningful deterioration in LV thickness (cardiac amyloid infiltration), LV systolic and diastolic function, and conduction during the 12-month tafamidis treatment period. Taken together, with findings from an earlier study in patients with TTR amyloidosis and Val30Met mutation, the current study provides evidence for the overall and cardiac safety of tafamidis 20 mg once daily as a disease-modifying therapy for patients with hereditary TTR amyloidosis, regardless of genotype.
HRV Indices and Conductive Disorders
Electrocardiographic and Holter monitoring results and HRV indices indicated cardiac autonomic dysfunction in most patients at baseline. Notably, mean echocardiographic, electrocardiographic, and Holter monitoring parameters did not show clinically meaningful changes over the 12-month treatment period [23]. Furthermore, the incidence of treatment-emergent abnormalities was low to moderate, and the pattern of observed abnormalities was comparable with that at baseline and consistent with underlying cardiac morbidity. Intriguingly, the number of patients with normal HRV measures increased over the 12-month treatment period. It will be of interest to investigate whether the normalization in HRV indices is reproducible or if it was purely due to chance, and to prospectively test the hypothesis that tafamidis, which was designed to slow or halt disease progression by preventing de novo amyloid deposition, may be able to improve existing cardiac autonomic dysfunction.
Cardiac Infiltration and LV Function
Echocardiographic abnormalities were seen in all patients at baseline and are consistent with the presence of cardiac amyloid deposits and restrictive/hypertrophic cardiomyopathy. With the exception of four patients with an increase in IVS thickness ≥2 mm, no change in LV wall thickness occurred during 12 months of tafamidis treatment, suggesting that TTR amyloid deposition did not progress in the majority of patients. This is an important point as liver transplantation is known to fail to prevent (or, in certain cases, has been reported to accelerate) progressive cardiac TTR amyloid deposition when cardiac hypertrophy exists before surgery [25, 28, 13]. LV filling pressure estimated by echocardiography also remained stable during the study.
NT-proBNP is a sensitive indicator of increased intraventricular pressures in congestive heart failure and has prognostic value for disease progression and mortality [29]. Therefore, a growing body of evidence suggests that NT-proBNP is a good marker for cardiac impairment (LV function and cardiac hypertrophy) in patients with hereditary TTR amyloidosis [30, 31]. In the current analysis, NT-proBNP concentrations were markedly elevated at baseline, yet no further meaningful increases were reported in the vast majority of participants reported during the course of tafamidis treatment.
In contrast with NT-proBNP, troponin I levels were normal in all but one patient at baseline. This finding is in agreement with a previous report that NT-proBNP is a more sensitive marker for cardiomyopathy in TTR amyloidosis patients than troponin I [30]. However, a recent study in patients with the acquired form of TTR amyloidosis suggests that troponin I may be of greater prognostic value than NT-proBNP [32].
Taken together, elevated NT-proBNP concentrations and measures of LV thickness indicate that the current study population had significant cardiac abnormalities at baseline. The findings that only four patients had mild increases in LV wall thickness and that the majority of participants had no meaningful increase in NT-proBNP over time suggest that cardiac disease may have been stable over the 12-month period of tafamidis treatment. Disease progression rates in TTR cardiac amyloidosis are not well defined, and clinical features are known to be highly heterogeneous across TTR mutations. Although the limited data available suggests that a significant deterioration may be observed in LV wall thickness, LV mass, and NT-proBNP levels during a 12-month period [33, 34] without a matched negative control group, it is unknown whether these measures would have worsened in the current study population in the absence of tafamidis treatment. Consequently, the absence of relevant cardiac disease progression in this small sample may or may not relate to tafamidis. However, the descriptive safety, tolerability, and outcome results are encouraging and justify the further evaluation of this TTR stabilizer for the treatment of TTR cardiac amyloidosis.
The hypothesis that tafamidis may slow the progression of TTR cardiomyopathy in patients with hereditary non-V30M transthyretin amyloidosis now needs to be confirmed in a larger patient cohort. To this end, a 30-month, multicenter, phase 3, double-blind, placebo-controlled, randomized trial aiming to evaluate the efficacy, safety, and tolerability of tafamidis in approximately 400 subjects diagnosed with transthyretin cardiomyopathy is ongoing (clinical trial identifier: NCT01994889). The prospect of a disease-modifying therapy that may effectively ameliorate cardiac problems associated with transthyretin amyloidosis may encourage the inclusion this often overlooked condition in the differential diagnosis in patients with heart failure. Furthermore, we hope that our attempts to define disease progression criteria to assess whether cardiac amyloid infiltration and cardiac dysfunction progressed during tafamidis treatment will stimulate additional research to test and validate these and other suitable response criteria (e.g., stabilization of amyloid deposition assessed using histologic or magnetic resonance imaging techniques) for reliable measurement of the impact of tafamidis and other investigational agents on transthyretin cardiac amyloidosis.
Study Limitations
This study had limitations common to those studies involving rare diseases, including the low number of study subjects and the lack of a comparator patient group. The ensuing inability to apply statistical tests to analyze the data is a major limitation of this small, hypothesis-generating study. In addition, this study was of relatively short duration, with an open-label design, and some of the analyses described here were post hoc. Nevertheless, our findings are notable in demonstrating that tafamidis seemed to cause no harm in this population of patients with non-Val30Met and non-Val122Ile TTR amyloidosis, most of whom had electrocardiographic or echocardiographic abnormalities at baseline and that there was little progression of amyloid cardiomyopathy during the year-long course of treatment.
Abbreviations
- E/A:
-
Ratio of early (E) over late (atrial A) peak diastolic mitral inflow velocity
- E/e′:
-
Ratio of early peak diastolic mitral inflow velocity (E) over the early diastolic mitral annular velocity (e′)
- HRDB:
-
Heart rate response to deep breathing
- HRV:
-
Heart rate variability
- IVS:
-
Left ventricular interventricular septal thickness
- LV:
-
Left ventricular
- LVEF:
-
Left ventricular ejection fraction
- LVPW:
-
Left ventricular posterior wall thickness
- NT-proBNP:
-
N-terminal pro-hormone brain natriuretic peptide
- TTR:
-
Transthyretin
References
Rapezzi, C., Quarta, C. C., Riva, L., Longhi, S., Gallelli, I., Lorenzini, M., Ciliberti, P., Biagini, E., Salvi, F., & Branzi, A. (2010). Transthyretin-related amyloidoses and the heart: a clinical overview. Nature Reviews Cardiology, 7(7), 398–408. doi:10.1038/nrcardio.2010.67.
Plante-Bordeneuve, V., & Said, G. (2011). Familial amyloid polyneuropathy. Lancet Neurology, 10(12), 1086–1097. doi:10.1016/S1474-4422(11)70246-0.
Rapezzi, C., Quarta, C. C., Obici, L., Perfetto, F., Longhi, S., Salvi, F., Biagini, E., Lorenzini, M., Grigioni, F., Leone, O., Cappelli, F., Palladini, G., Rimessi, P., Ferlini, A., Arpesella, G., Pinna, A. D., Merlini, G., & Perlini, S. (2013). Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: an Italian perspective. European Heart Journal, 34(7), 520–528. doi:10.1093/eurheartj/ehs123.
Connors, L. H., Lim, A., Prokaeva, T., Roskens, V. A., & Costello, C. E. (2003). Tabulation of human transthyretin (TTR) variants, 2003. Amyloid, 10(3), 160–184. doi:10.3109/13506120308998998.
Andrade, C., Araki, S., Block, W. D., Cohen, A. S., Jackson, C. E., Kuroiwa, Y., Nissim, J., Sohar, E., McKusick, V. A., & Van Allen, M. W. (1970). Hereditary amyloidosis. Arthritis and Rheumatism, 13(6), 902–915.
Connors, L. H., Prokaeva, T., Lim, A., Théberge, R., Falk, R. H., Doros, G., Berg, A., Costello, C. E., O’Hara, C., Seldin, D. C., & Skinner, M. (2009). Cardiac amyloidosis in African Americans: comparison of clinical and laboratory features of transthyretin V122I amyloidosis and immunoglobulin light chain amyloidosis. American Heart Journal, 158(4), 607–614. doi:10.1016/j.ahj.2009.08.006.
Eriksson, P., Karp, K., Bjerle, P., & Olofsson, B. O. (1984). Disturbances of cardiac rhythm and conduction in familial amyloidosis with polyneuropathy. British Heart Journal, 51(6), 658–662.
Rapezzi, C., Perugini, E., Salvi, F., Grigioni, F., Riva, L., Cooke, R. M., Ferlini, A., Rimessi, P., Bacchi-Reggiani, L., Ciliberti, P., Pastorelli, F., Leone, O., Bartolomei, I., Pinna, A. D., Arpesella, G., & Branzi, A. (2006). Phenotypic and genotypic heterogeneity in transthyretin-related cardiac amyloidosis: towards tailoring of therapeutic strategies? Amyloid, 13(3), 143–153. doi:10.1080/13506120600877136.
Jacobson, D. R., Pastore, R. D., Yaghoubian, R., Kane, I., Gallo, G., Buck, F. S., & Buxbaum, J. N. (1997). Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. New England Journal of Medicine, 336(7), 466–473. doi:10.1056/NEJM199702133360703.
Herlenius, G., Wilczek, H. E., Larsson, M., & Ericzon, B. G. (2004). Ten years of international experience with liver transplantation for familial amyloidotic polyneuropathy: results from the Familial Amyloidotic Polyneuropathy World Transplant Registry. Transplantation, 77(1), 64–71. doi:10.1097/01.TP.0000092307.98347.CB.
Okamoto, S., Wixner, J., Obayashi, K., Ando, Y., Ericzon, B. G., Friman, S., Uchino, M., & Suhr, O. B. (2009). Liver transplantation for familial amyloidotic polyneuropathy: impact on Swedish patients’ survival. Liver Transplantation, 15(10), 1229–1235. doi:10.1002/lt.21817.
Yamamoto, S., Wilczek, H. E., Nowak, G., Larsson, M., Oksanen, A., Iwata, T., Gjertsen, H., Söderdahl, G., Wikström, L., Ando, Y., Suhr, O. B., & Ericzon, B. G. (2007). Liver transplantation for familial amyloidotic polyneuropathy (FAP): a single-center experience over 16 years. American Journal of Transplantation, 7(11), 2597–2604. doi:10.1111/j.1600-6143.2007.01969.x.
Stangou, A. J., Hawkins, P. N., Heaton, N. D., Rela, M., Monaghan, M., Nihoyannopoulos, P., O’Grady, J., Pepys, M. B., & Williams, R. (1998). Progressive cardiac amyloidosis following liver transplantation for familial amyloid polyneuropathy: implications for amyloid fibrillogenesis. Transplantation, 66(2), 229–233.
Hörnsten, R., Wiklund, U., Olofsson, B. O., Jensen, S. M., & Suhr, O. B. (2004). Liver transplantation does not prevent the development of life-threatening arrhythmia in familial amyloidotic polyneuropathy, Portuguese-type (ATTR Val30Met) patients. Transplantation, 78(1), 112–116.
Yazaki, M., Tokuda, T., Nakamura, A., Higashikata, T., Koyama, J., Higuchi, K., Harihara, Y., Baba, S., Kametani, F., & Ikeda, S. (2000). Cardiac amyloid in patients with familial amyloid polyneuropathy consists of abundant wild-type transthyretin. Biochemical and Biophysical Research Communications, 274(3), 702–706. doi:10.1006/bbrc.2000.3203.
Wilczek, H. E., Larsson, M., & Ericzon, B. G. (2011). Long-term data from the Familial Amyloidotic Polyneuropathy World Transplant Registry (FAPWTR). Amyloid, 18(Suppl 1), 193–195. doi:10.3109/13506129.2011.574354072.
Hou, X., Aguilar, M. I., & Small, D. H. (2007). Transthyretin and familial amyloidotic polyneuropathy. Recent progress in understanding the molecular mechanism of neurodegeneration. FEBS Journal, 274(7), 1637–1650. doi:10.1111/j.1742-4658.2007.05712.x.
Sekijima, Y., Kelly, J. W., & Ikeda, S. (2008). Pathogenesis of and therapeutic strategies to ameliorate the transthyretin amyloidoses. Current Pharmaceutical Design, 14(30), 3219–3230.
Johnson, S. M., Wiseman, R. L., Sekijima, Y., Green, N. S., Adamski-Werner, S. L., & Kelly, J. W. (2005). Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: a focus on the transthyretin amyloidoses. Accounts of Chemical Research, 38(12), 911–921. doi:10.1021/ar020073i.
Berk, J. L., Suhr, O. B., Obici, L., Sekijima, Y., Zeldenrust, S. R., Yamashita, T., Heneghan, M. A., Gorevic, P. D., Litchy, W. J., Wiesman, J. F., Nordh, E., Corato, M., Lozza, A., Cortese, A., Robinson-Papp, J., Colton, T., Rybin, D. V., Bisbee, A. B., Ando, Y., Ikeda, S., Seldin, D. C., Merlini, G., Skinner, M., Kelly, J. W., & Dyck, P. J. (2013). Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA, 310(24), 2658–2667. doi:10.1001/jama.2013.283815.
Bulawa, C. E., Connelly, S., Devit, M., Wang, L., Weigel, C., Fleming, J. A., Packman, J., Powers, E. T., Wiseman, R. L., Foss, T. R., Wilson, I. A., Kelly, J. W., & Labaudinière, R. (2012). Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proceedings of the National Academy of Sciences of the United States of America, 109(24), 9629–9634. doi:10.1073/pnas.1121005109.
Coelho, T., Maia, L. F., Martins da Silva, A., Waddington Cruz, M., Planté-Bordeneuve, V., Lozeron, P., Suhr, O. B., Campistol, J. M., Conceição, I. M., Schmidt, H. H., Trigo, P., Kelly, J. W., Labaudinière, R., Chan, J., Packman, J., Wilson, A., & Grogan, D. R. (2012). Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology, 79(8), 785–792. doi:10.1212/WNL.0b013e3182661eb1.
Merlini, G., Planté-Bordeneuve, V., Judge, D. P., Schmidt, H., Obici, L., Perlini, S., Packman, J., Tripp, T., & Grogan, D. R. (2013). Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. Journal of Cardiovascular Translational Research, 6(6), 1011–1020. doi:10.1007/s12265-013-9512-x.
European Medicines Agency (2013) EMA/295794/2013—European public assessment report (EPAR) for Vyndaqel.
Dubrey, S. W., Davidoff, R., Skinner, M., Bergethon, P., Lewis, D., & Falk, R. H. (1997). Progression of ventricular wall thickening after liver transplantation for familial amyloidosis. Transplantation, 64(1), 74–80.
Gertz, M. A., Comenzo, R., Falk, R. H., Fermand, J. P., Hazenberg, B. P., Hawkins, P. N., Merlini, G., Moreau, P., Ronco, P., Sanchorawala, V., Sezer, O., Solomon, A., & Grateau, G. (2005). Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18-22 April 2004. American Journal of Hematology, 79(4), 319–328. doi:10.1002/ajh.20381.
McMurray, J. J., Adamopoulos, S., Anker, S. D., Auricchio, A., Böhm, M., Dickstein, K., Falk, V., Filippatos, G., Fonseca, C., Gomez-Sanchez, M. A., Jaarsma, T., Køber, L., Lip, G. Y., Maggioni, A. P., Parkhomenko, A., Pieske, B. M., Popescu, B. A., Rønnevik, P. K., Rutten, F. H., Schwitter, J., Seferovic, P., Stepinska, J., Trindade, P. T., Voors, A. A., Zannad, F., & Zeiher, A. (2012). ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: the task force for the diagnosis and treatment of acute and chronic heart failure 2012 of the European Society of Cardiology. developed in collaboration with the Heart Failure Association (HFA) of the ESC. European Heart Journal, 33(14), 1787–1847. doi:10.1093/eurheartj/ehs104.
Pomfret, E. A., Lewis, W. D., Jenkins, R. L., Bergethon, P., Dubrey, S. W., Reisinger, J., Falk, R. H., & Skinner, M. (1998). Effect of orthotopic liver transplantation on the progression of familial amyloidotic polyneuropathy. Transplantation, 65(7), 918–925.
Bhardwaj, A., Rehman, S. U., Mohammed, A., Baggish, A. L., Moore, S. A., & Januzzi, J. L., Jr. (2010). Design and methods of the Pro-B Type Natriuretic Peptide Outpatient Tailored Chronic Heart Failure Therapy (PROTECT) Study. American Heart Journal, 159(4), 532–538. doi:10.1016/j.ahj.2010.01.005. e531.
Suhr, O. B., Anan, I., Backman, C., Karlsson, A., Lindqvist, P., Morner, S., & Waldenstrom, A. (2008). Do troponin and B-natriuretic peptide detect cardiomyopathy in transthyretin amyloidosis? Journal of Internal Medicine, 263(3), 294–301. doi:10.1111/j.1365-2796.2007.01888.x.
Damy, T., Deux, J. F., Moutereau, S., Guendouz, S., Mohty, D., Rappeneau, S., Guellich, A., Hittinger, L., Loric, S., Lefaucheur, J. P., & Plante-Bordeneuve, V. (2013). Role of natriuretic peptide to predict cardiac abnormalities in patients with hereditary transthyretin amyloidosis. Amyloid, 20(4), 212–220. doi:10.3109/13506129.2013.825240.
Pinney, J. H., Whelan, C. J., Petrie, A., Dungu, J., Banypersad, S. M., Sattianayagam, P., Wechalekar, A., Gibbs, S. D., Venner, C. P., Wassef, N., McCarthy, C. A., Gilbertson, J. A., Rowczenio, D., Hawkins, P. N., Gillmore, J. D., & Lachmann, H. J. (2013). Senile systemic amyloidosis: clinical features at presentation and outcome. Journal of American Heart Association, 2(2), e000098. doi:10.1161/JAHA.113.000098.
Dyck, P. J., Litchy, W. J., Lehman, K. A., Hokanson, J. L., Low, P. A., & O’Brien, P. C. (1995). Variables influencing neuropathic endpoints: the Rochester diabetic neuropathy study of healthy subjects. Neurology, 45(6), 1115–1121.
Dyck, P. J., O’Brien, P. C., Litchy, W. J., Harper, C. M., & Klein, C. J. (2005). Monotonicity of nerve tests in diabetes: subclinical nerve dysfunction precedes diagnosis of polyneuropathy. Diabetes Care, 28(9), 2192–2200.
Vinik, E. J., Vinik, A. I., Paulson, J. F., Merkies, I. S., Packman, J., Grogan, D. R., & Coelho, T. (2014). Norfolk QOL-DN: validation of a patient reported outcome measure in transthyretin familial amyloid polyneuropathy. Journal of the Peripheral Nervous System, 19(2), 104–114. doi:10.1111/jns5.12059.
Sources of Funding
This study was funded by Pfizer Inc. Medical writing support for this manuscript was provided by Susanne Vidot, Ph.D. of Engage Scientific Solutions and was funded by Pfizer Inc.
Conflict of Interest
Drs. Damy, Judge, and Kristen have received research support from and served on advisory boards for Pfizer Inc. Drs. Berthet, Aarts, and Li are employees of and hold stock options in Pfizer Inc.
Ethics Statement
The study was conducted in accordance with the Declaration of Helsinki and with the ethical standards of the responsible committee on human experimentation (institutional and national) and was approved by the relevant ethics committee at each site. All patients provided written informed consent. No animal studies were carried out by the authors for this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor-in-Chief Jennifer L. Hall oversaw the review of this article
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Damy, T., Judge, D.P., Kristen, A.V. et al. Cardiac Findings and Events Observed in an Open-Label Clinical Trial of Tafamidis in Patients with non-Val30Met and non-Val122Ile Hereditary Transthyretin Amyloidosis. J. of Cardiovasc. Trans. Res. 8, 117–127 (2015). https://doi.org/10.1007/s12265-015-9613-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-015-9613-9